




DEGENERAZIONI TAPPETO RETINICHE
Degenreazioni retiniche: un vasto gruppo di malattie
DEGENERAZIONI TAPPETO RETINICHE
retinitepigmentosa
Le degenerazioni tappeto-retiniche sono un vasto gruppo di malattie, variamente trasmesse dal punto di vista genetico(Deutman, 1971) con modalità autosomiche ed eterosomiche, dominanti o recessive, con localizzazione retinica centrale, periferica o mista,per lo più caratterizzate da progressive alterazioni funzionali (del visus, campimetriche, cromatiche, adattometriche),elettrofunzionali(elettrooculografiche, elettroretinografiche, dei potenziali visivi evocati) e naturalmente oftalmoscopiche nonché fluorangiografiche.
Fra le più note di queste malattie vogliamo qui ricordare la retinopatia pigmentaria (nelle sue varianti: con o senza pigmento, uni o bilaterale,completa o a settore, punteggiata albescente);
tra le forme a localizzazione prevalentemente periferica, la distrofia dei coni e la malattia di Stargardt; tra le forme a localizzazione centrale,il fundus fl avimaculatus e la distrofia dei coni edei bastoncelli fra le forme miste.
A tutt'oggi non sono ancora stati chiariti i meccanismi eziopatogenetici delle eredo-distrofie per le quali tuttavia si ipotizza per lo più un danno di natura abiotrofica a carico di uno o più strati retino-coroidali- SIRAVO (Wirth
Cavallacci, 1984); il neuroepitelio nella malattia di Stargardt, nella distrofia progressiva dei coni, nella retìnopatia pigmentaria e l'epitelio pigmentato nel fundus flavimaculatus (Deutman,1971).
Per la retinopatia pigmentosa, inoltre, è stata da taluni (Heredia e Coll., 1984) ipotizzata una patogenesi autoimmunitaria.
L'intero gruppo di queste patologie, rappresenta un'importante causa di ipovedenza nella popolazione mondiale, data anche la scarsità, per non dire quasi l'assenza, di presidi terapeutici realmente efficaci finora a nostra disposizione
Si riconoscono otto diverse forme:
1. amaurosi congenita di Leber
2. distrofia dei coni
3. distrofia ialina della retina
4. distrofia vitelliforme di Best
5. distrofia vitreoretinica
6. malattia di Stargardt
7. retinite pigmentosa
retinitepigmentosa
8. retinite puntata albescens
AMAUROSI CONGENITA DI LEBER
AMAUROSI CONGENITA DI LEBER TERAPIA GENICA
L'amaurosi congenita di Leber è una malattia genetica che colpisce la retina, provocando cecità o grave danneggiamento della vista fin dalla infanzia (in genere l'esordio è nei primi sei mesi di vita). È la causa più frequente di cecità infantile ereditaria, con un'incidenza di 3 casi ogni 100.000 nati vivi. Oltre alla marcata ipovisione, un altro sintomo tipico è il nistagmo, cioè il movimento continuo degli occhi.
Si conoscono 10-12 geni associati (quando alterati) all'amaurosi congenita di Leber; nel 10 per cento dei casi la malattia è causata da mutazioni del gene RPE65. La trasmissione avviene con modalità autosomica recessiva: perché la malattia si manifesti occorre ereditare le due copie alterate del gene coinvolto da entrambi i genitori.
Terapia genica nell’amaurosi congenita di Leber con mutazioni nel gene RPE65
La terapia genica è potenzialmente in grado di revertire la malattia o prevenire ulteriore deterioramento della visione in pazienti con degenerazione retinica ereditaria e incurabile.
Uno studio di fase I ha valutato l’effetto della terapia genica sulla funzione della retina e sulla funzione visiva in bambini e adulti con amaurosi congenita di Leber.
Sono state valutate la funzione della retina e quella visiva in 12 pazienti di età compresa tra 8 e 44 anni con amaurosi congenita di Leber associata a RPE65, sottoposti a una iniezione sottoretinica di virus adeno-associato contenente un gene che codifica per una proteina necessaria per l’attività isomeroidrolasica dell’epitelio pigmentato retinico ( AAV2-hRPE65v2 ) nell’occhio in condizioni peggiori a bassa [ 1.5 x 10(10) vettori ], media [ 4.8 x 10(10) vettori ] o alta dose [ 1,5 x 10(11) vettori ] fino a 2 anni.
AAV2-hRPE65v2 è risultato ben tollerato e tutti i pazienti hanno mostrato un miglioramento sostenuto nelle valutazioni soggettive e oggettive della visione ( adattometria al buio, pupillometria, elettroretinografia, nistagmo ).
I pazienti hanno mostrato un incremento di almeno 2 unità logaritmiche nella risposta della pupilla alla luce, e un bambino di 8 anni ha raggiunto circa lo stesso livello di sensibilità alla luce dei suoi coetanei senza problemi di vista.
Il miglioramento maggiore è stato osservato nei bambini: tutti hanno acquisito ambulatory vision ( percezione delle ombre ).
Dallo studio è emerso che la sicurezza, il grado e la stabilità del miglioramento della visione in tutti i pazienti sono a sostegno dell’uso della terapia genica mediata da virus adeno-associati per il trattamento di disturbi ereditari della retina.
Gli interventi più precoci sono associati a risultati migliori. ( Xagena2009 )
Nuove speranze per sconfiggere la cecità e recuperare parzialmente la vista. Una ricerca pubblicata sul New England Journal of Medicine, ha scoperto una cura per l’amaurosi congenita di Leber, una forma di cecità ereditaria. Tre italiani che ne sono affetti sono già stati operati e hanno avuto dei grandi miglioramenti nella visione, mentre è già iniziato il trattamento per un quarto paziente.
Per arrivare a questa terapia, è stato necessario il lavoro congiunto di ricercatori italiani coordinati dal Children Hospital di Filadelfia e dall’Istituto Telethon di Genetica e Medicina (Tigem) e dal Dipartimento di Oftalmologia della Seconda Università degli Studi di Napoli.
La terapia genica consiste nell’iniettare, nello spazio sottoretinico dell’occhio dei pazienti, un vettore virale con la versione sana del gene alterato. Il gene corretto provvede poi a produrre la proteina mancante nei non vedenti affetti da amaurosi congenita.
Come è accaduto ai pazienti sottoposti a trattamento, una ragazza di 19 anni di Pavia, e due gemelli siciliani. I tre ora riescono ad effettuare percorsi ad ostacoli e hanno una visibilità nettamente migliorata.
Maguire AM et al, Lancet 2009; 374: 1597-1605
DISTROFIA DEI CONI
Sinonimi:
·
Degenerazione combinata coni-bastoncelli
·
Degenerazione progressiva coni-bastoncelli
·
Distrofia coni-bastoncelli
·
Distrofia retinica coni- bastoncelli
· Distrofia retinica dei coni
E’ caratterizzata da degenerazione di coni e bastoncelli e dalla conseguente alterazione della capacità visiva.
Le distrofie dei coni e dei bastoncelli (CRD) sono distrofie retiniche ereditarie, che appartengono al gruppo delle retiniti pigmentose. La prevalenza è stimata a 1:40.000. Le CRD sono caratterizzate da depositi di pigmento sulla retina, visibili all'esame del fondo dell'occhio, localizzati soprattutto nella regione maculare. A differenza della classica retinite pigmentosa (RP), nota anche come distrofia dei bastoncelli e dei coni (RCD), dovuta alla perdita primitiva dei fotorecettori dei bastoncelli e, secondariamente, dei fotorecettori dei coni, le CRD sono caratterizzate da una sequenza opposta. La CRD presenta un coinvolgimento primitivo dei coni e, a volte, la perdita concomitante dei coni e dei bastoncelli. Questo spiega il fatto che i sintomi della CRD consistano essenzialmente in una riduzione dell'acuità visiva, difetti della visione dei colori, fotofobia e diminuzione della sensibilità visiva centrale, seguita successivamente dalla perdita progressiva della visione periferica e dalla cecità notturna. Il decorso clinico della CRD è di solito più grave e rapido rispetto a quello della RCD, in quanto può comportare cecità precoce e invalidità. Tuttavia, nello stadio finale, le CRD non si differenziano dalle RCD. Le CRD sono spesso non-sindromiche, ma possono comparire all'interno di alcune sindromi, come la sindrome di Bardet-Biedl (si veda questo termine) e l'Atassia Spinocerebellare tipo 7 (SCA7). Le CRD non-sindromiche sono geneticamente eterogenee (finora sono stati identificati dieci geni clonati e tre loci). I quattro principali geni-malattia coinvolti nella patogenesi delle CRD sono ABCA4 (che causa la malattia di Stargardt, si veda questo termine, e il 30-60% delle CRD autosomiche recessive), CRX e GUCY2D (responsabili di numerose forme di CDR autosomiche dominanti) e RPGR (che causa circa i 2/3 delle RP legate all'X e anche di una percentuale sottostimata delle CRD legate all'X). È probabile che alcune mutazioni nei geni che causano le RP e le distrofie maculari siano responsabili anche delle CRD. La diagnosi della CRD si basa sulla storia clinica, l'esame del fondo e l'elettroretinogramma. La diagnosi molecolare è disponibile per alcuni geni e la consulenza genetica è sempre indicata. Al momento non è disponibile nessuna terapia in grado di arrestare l'evoluzione della malattia o di restituire la vista. La prognosi per la vista è negativa. La terapia ha lo scopo di rallentare il processo degenerativo, trattare le complicazioni e aiutare i pazienti a fare fronte all'impatto psico-sociale della cecità. *Autore: Dott. C. Hamel (Febbraio 20007)*. Tratto da Cone rod dystrophies. Orphanet J Rare Dis. 2007;2:7.
L'amelogenesi imperfetta (AI) identifica un gruppo di malattie ereditarie dei denti, accomunate da un'anomalia dello smalto, che può essere sottile ma normale, oppure ipomineralizzato, o entrambi. Nella AI sono state descritte tutte le modalità di trasmissione. Tra le forme sindromiche di AI è stata osservata un'associazione con la distrofia retinica dei coni e dei bastoncelli, una malattia retinica rara che causa perdita iniziale della visione centrale, della visione cromatica e fotofobia, prima dei 10 anni, e successiva cecità notturna e restringimento del campo visivo. Sono state descritte diverse dorme di trasmissione, sia per l'AI che per la distrofia, mentre la sindrome che associa entrambi i sintomi è stata descritta in un'unica famiglia con 29 persone affette, a trasmissione autosomica recessiva.
DISTROFIA IALINA DELLA RETINA
Sinonimi:
·
Degenerazione ialoido-retinica di Favre
·
Degenerazione ialoide-tappeto-retinica
·
Degenerazione microfibrillare vitreoretinica di Favre
· Malattia di Golman-Favre
Malattia caratterizzata da perdita graduale della vista o cecità notturna e da segni oculari che comprendono la liquefazione del corpo vitreo, la retinoschisi maculare e l’atrofia e la pigmentazione periferica dell’epitelio pigmentato della retina.
L’elettroretinogramma è estinto o marcatamente anomalo.
Origini e diffusione
E’ una malattia rara a prevalenza non nota. Maschi e femmine sono affetti in eguale misura.I pazienti giungono all’osservazione clinica nel corso delle prime duedecadi di vita per una cecità notturna associata a retinopatia pigmentaria insidiosa spesso bilaterale. Possono evidenziarsi retinoschisi periferica e ampie zone di degenerazione tipo lattice che provocano lacune retiniche.
Segni oculari associati possono essere la liquefazione del corpovitreo e la presenza di membrane epiretiniche. I pazienti possono sviluppare precocemente cataratta e distacco di retina. I vasi periferici retinici appaiono opachi o sclerotici. L’elettroretinogramma è estinto o marcatamente alterato già nei primi anni di vita. L’acuità visiva rimane relativamente stabile nelle prime due decadi, ma più tardivamente mostra un peggioramento significativo.
E’ una malattia genetica a trasmissione autosomica recessiva. Il gene responsabile non è noto.Criteri diagnosticiLa diagnosi rimane clinica e strumentale. Non esiste al momento un test genetico di conferma diagnostica.
Diagnosi differenziale
E’ necessario differenziare dalle altre forme di distrofia retinica:amaurosi congenita di Leber; distrofia vitelliforme di Best; distrofia vitreoretinica; retinite pigmentosa; retinite puntata albescens;malattia di stargardt.
Terapia
Non esiste al momento alcun trattamento causale della malattia. E’possibile effettuare un trattamento profilattico delle rotture retiniche asintomatiche per prevenire il distacco di retina,come nel suo caso.
E' una malattia genetica a trasmissione autosomica recessiva ,come lei diceva,ma non possono essere effettuati studi genetici specifici perchè non è conosciuto il gene responsabile!!!!
DISTROFIA VITELLIFORME DI BEST
Sinonimi:
·
Cisti congenita vitelliforme della macula
·
Degenerazione maculare vitelliforme
·
Distacco centrale essudativo della retina
·
Distrofia cistoide centrale
· Distrofia maculare atipica vitelliforme
E’ un’affezione maculare bilaterale ereditaria dominante che presenta una penetranza incompleta ed espressività variabile. Può essere osservata anche in neonati, ma è più frequente nei giovani adulti. La diagnosi è spesso agevole e si avvale sia dell’aspetto clinico, fluorangiografico e anche gli esami elettrofisiologici, soprattutto l’elettoculogramma. Solitamente il visus è ben conservato e solo nelle forme più evolute si può avere un grado visivo piuttosto basso.
Le manifestazioni cliniche sono estremamente variabili sia per l’aspetto delle lesioni che per l’età di insorgenza. A secondo dell’aspetto della lesione sono stati descritti 5 stadi (classificazione di Mohler & Fine, 1981).
STADIO
ASPETTO DELLA MACULA
0 fondo normale
1 alterazioni minori dell’epitelio pigmentato retinico
2 tipica lesione vitelliforme
2a aspetto a "uovo strapazzato"
3 fase dello "pseudoipopion"
4a epitelio pigmentato retinico atrofico
4b tessuto cicatriziale fibroso
4c neovascolarizzazione coroideale
Esistono delle varianti della malattia di Best caratterizzate da:
Forma unilaterale: identica alla forma già descritta ma che interessa un solo occhio.
Forme multiple: esiste una lesione maculare classica associata ad altre lesioni più piccole che si trovano in stadi evolutivi più precoci, ma che evolvono ugualmente verso la cicatrice centrale.
Forma paramaculare: la lesione è spostata temporalmente sfiorando la foveola che rimane intatta. In questi casi l’acuità visiva rimane sempre buona.
Forma essudativa neovascolare: si ha un improvviso abbassamento dell’acuità visiva e in alcuni casi può essere effettuato un trattamento laser.
DISTROFIA VITREORETINICA
Sinonimi:
·
Cisti gigante della retina
·
Cisti retinica congenita
·
Retinoschisi della fovea
· Retinoschisi giovanile X-linked
La distrofia vitreoretinica è una malattia che colpisce quasi esclusivamente il sesso maschile. Sebbene la condizione abbia un esordio patogenetico congenito, i sintomi diventano evidenti solo verso i dieci anni.
Il deficit visivo è dovuto a una fenditura della retina che si divide in due strati.
Tale fenditura riguarda principalmente la macula, porzione centrale della retina responsabile della visione fine e dettagliata e della discriminazione dei colori. All’esame fundoscopico si riscontrano a
livello della fovea strisce raggiformi. Gli spazi creati dalla fenditura
retinica sono spesso occupati da vescicole e da vasi sanguigni, che vanno incontro a rottura con conseguente sanguinamento nel corpo vitreo.
MALATTIA DI STARGARDT
Sinonimi:
·
Degenerazione maculare giovanile
·
Distrofia maculare pura
· Fundus flavimaculato con degenerazione maculare
La degenerazione ereditaria maculare giovanile di Stargardt
Sinonimo della Degenerazione maculare giovanile o Distrofia maculare pura
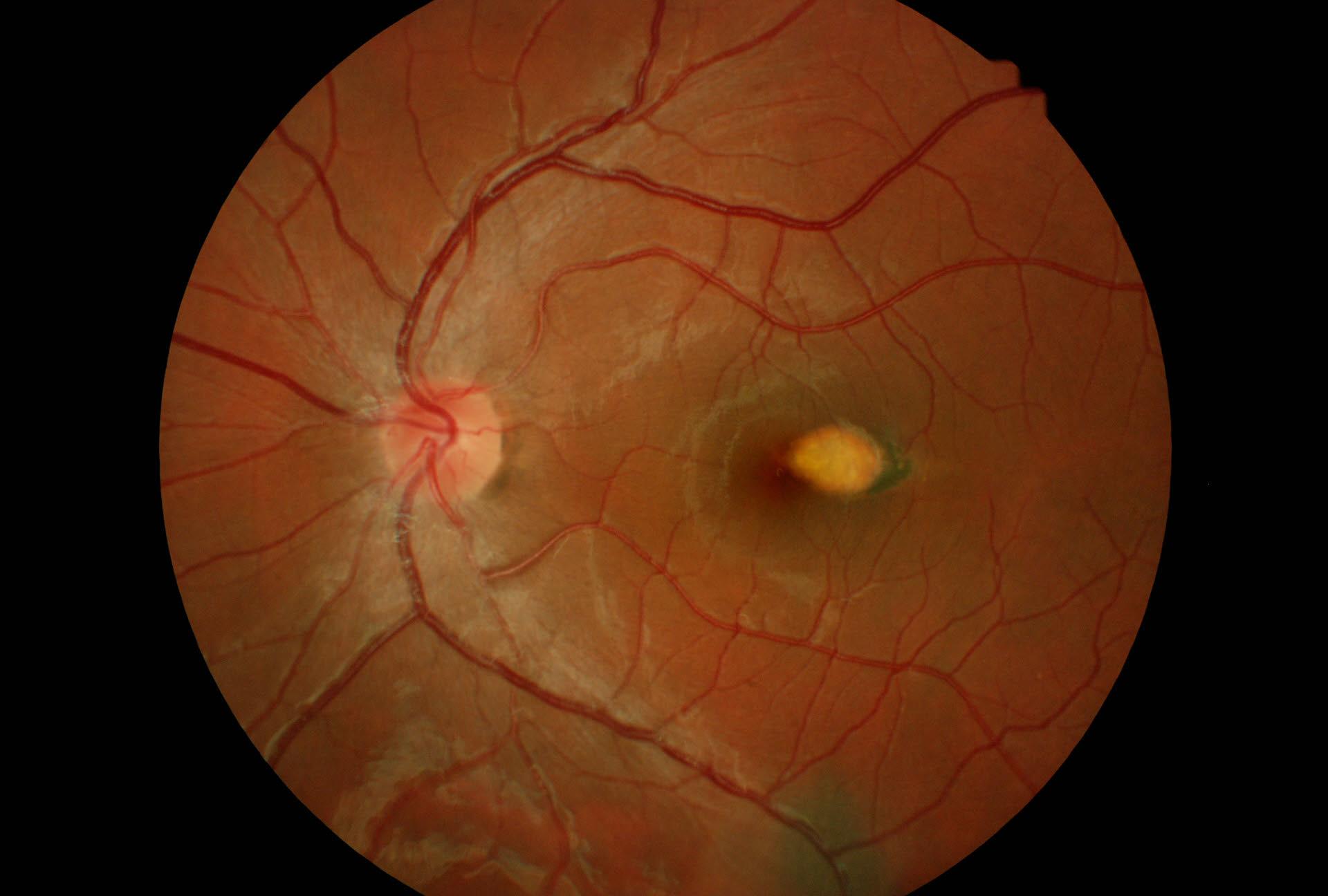
La malattia di Stargardt è una forma giovanile di degenerazione ereditaria della macula, caratterizzata da macchie giallastre rotonde o pisciformi attorno alla macula, a livello dell'epitelio pigmentato della retina (EPR).
La malattia di Stargardt è la forma più comune di distrofia ereditaria della macula, con una prevalenza di circa 1:8.000-10.000.
La malattia insorge caratteristicamente nella prima o nella seconda decade di vita e si manifesta con una riduzione dell'acuità visiva.
Negli stadi precoci, la macula mostra di solito evidenti modificazioni dell'epitelio pigmentato della retina, che sono seguiti dalla comparsa di una zona ovoidale orizzontale di atrofia "bronzea".
Negli ultimi stadi, le lesioni della macula possono associarsi ad una distrofia areolare centrale della coroide. L'angiografia con fluorescina rivela una caratteristica coroide scura ("silenzio coroideo"), probabilmente dovuta all'accumulo di lipofuscina nell'epitelio pigmentato della retina. Questa malattia ha di solito un'eredità autosomica recessiva, ma sono state descritte alcune famiglie dominanti. La forma autosomica è dovuta a mutazioni nel gene ABCR, che codifica per una proteina del trasporto attraverso la membrana cellulare, espressa nei segmenti esterni dei bastoncelli.
Non è al momento disponibile nessuna terapia efficace per la malattia di Stargardt,ma ci stiamo occupando di tutto quanto la ricerca propone!!!!
RETINITE PIGMENTOSA
retinitepigmentosa
RETINITE PUNTATA ALBESCENTE
Sinonimi:
· Retinopatia punteggiata albescente
La retinite puntata albescens si caratterizza per la presenza regolare su tutto il territorio retinico di chiazzette bianche, che possono precedere o coesistere con la pigmentazione tipica della retinite pigmentosa. I sintomi peculiari sono l’emeralopia e il difetto
campimetrico, caratterizzato inizialmente da uno scotoma centrale e,successivamente, da un restringimento concentrico che comporta
una visione tubolare.
L’ERG è alterato o estinto.
Malattia genetica a trasmissione autosomica recessiva. Il gene responsabile non è ancora noto.
Un caro saluto
Prof.Duilio Siravo
siravo@supereva.it
http://drsiravoduilio.beepworld.it
Cell.:3385710585
PROF.DOTT. DUILIO SIRAVO
http://drsiravoduilio.beepworld.it





{kind=link}