Malattia di Behçet
Malattia di Behçet
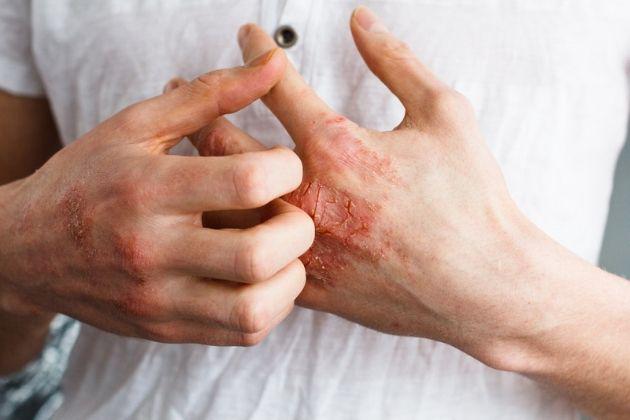
La sindrome di Behçet è un disordine infiammatorio multisistemico recidivante ad eziologia sconosciuta caratterizzato da afte orali, genitali, uveite e che frequentemente coinvolge le articolazioni, la cute, il sistema nervoso centrale e il tratto gastrointestinale.
In Italia l'incidenza è di 2,4/milione di abitanti e la prevalenza di 3,8/100000. Esiste una associazione genetica con l'antigene HLA-B51 e nella razza caucasica dell'antigene HLA-B57. Anatomo patologicamente si osservano lesioni più importanti anche se non specifiche a carico dei piccoli vasi. Le pareti di queste sono infiltrate da cellule linfomonocitarie talora con aree di necrosi fibrinoide. Si possono trovare trombosi venose e meno frequentemente arteriose.
QUADRO CLINICO
Lesioni aftose nel cavo orale con frequenti recidive (almeno 3 volte in una anno).
Appaiono singole o multiple, dolenti di morfologia rotondeggiante con margine eritematoso, coperte da pseudomembrane grigio-biancastre o con bse fibrinosa giallastra. La localizzazione anteriore è più frequente. In relazione all'aspetto vengono classificate in minor, major e erpetiformi. Ulcere genitali nell'80-94% appaiono a livello dello scroto o della vuvla e spesso più ampie e profonde di quelle orali. Altre lesioni si estrinsecano in oltre l'80% dei casi (eritema nodoso, eruzione vescicolo-papulo-pustolose). Interessamento oculare nel 20% dei casi all'esordio e nel 50% dei pazienti durante la malattia ed è caratterizzata da iridociclite unilaterale (20%) o bilaterale (80%), ipopion (raccolta di pus nella camera anteriore dell'occhio) o uveite o panuveite. La classificazione dell'uveite come anteriore o posteriore è importante dal punto di vista diagnostico e terapeutico in quanto il coinvolgimento della porzione posteriore è indice di cronicità-persistenza ed è associato a riduzione del visus preogressiva e cumolativa, che se non trattata porterà alla cecità. Gli attacchi di uveite anteriori, spesso bilaterali si manifestano come episodi di dolore retroorbitario, fotofobia, riduzione lieve o moderata del visus, miosi reattiva, lacrimazione. La vasodilatazione congiuntivale acuta di tipo ciliale si evidenzia come un disco di iperemia cirumcorneale con alone violaceo (anastomosi vascolari ciliari e sclero congiuntivale). Esame alla lampada a fessura, numerosi linfociti fluttanti nella camera anteriore dell'occhio che hanno attraversato l'endotelio danneggiato. Eziologia e patogenesi -elemento genetico (HLA-B51 e B57) - presenza di anticorpi anti-saccharomyces cereviasiae. - antigeni implicati (antigene retinico S, tropomiosina, recettore delle cellule NK, alfa-enolasi riconosciuta da anticorpi anti-endotelio, AECA). Diagnosi - Afte orali ricorrenti (recidivanti almeno 3 volta in un anno) + almeno 2 delle seguenti: - Afte genitali - Lesioni oculari - Pathergy test positivo. Terapia: Spesso i farmaci sono usati in combinazione per migliorare l'efficacia e ridurre gli effetti collaterali. Le afte orali e genitali rispondono spesso all'applicazione topica di glucocorticoidi ed alla somministrazione di colchina. Ottimo anche il sucralfato per via topica. L'uso di glucocorticoidi per via sistemica è riservata alle esacerbazioni acute, incluso le manifestazioni oculari e nuerologiche. Spesso usati in associazione con farmaci immunosoppressori come gli inibitori della calcineurina (ciclosporina, tacrolimus) e l'azatioprina. L'uso della ciclofosfamide è riservato alle forme refrattarie di vasculite. L'uso di agenti anti-TNF-alfa in tutte le forma di manifestazioni hanno beneficiato di tale trattamento.
La malattia di Behçet è una vasculite sistemica (infiammazione dei vasi sanguigni) coinvolgente le arterie e le vene di vario calibro che, oltre a causare ulcere orali e genitali ricorrenti e lesioni oculari, può causare vari tipi di lesioni cutanee, artrite, trombosi venosa profonda(TVP), tromboflebiti, infiammazione intestinale e del sistema nervoso centrale. La malattia di Behçet è un disordine raro e cronico di origine autoimmune e solitamente inizia nella fascia della terza decade di età (20-30 anni) anche se sono colpiti gruppi di tutte le età (compresa l'età infantile). È diffusa in quasi tutti i paesi del mondo anche se la maggior incidenza è soprattutto nella aree che vanno dal bacino del Mediterraneo al Giappone seguendo la vecchia via della seta.
A tutt'oggi non sono note le cause della malattia, si sa per certo che non è infettiva o contagiosa né la trasmissione avviene per via sessuale. È stata accertata una predisposizione genetica: nei soggetti affetti si riscontra infatti, con maggior frequenza rispetto alla popolazione generale, un particolare antigene di istocompatibilità (HLA-B51), e la sua presenza si associa ad un rischio maggiore di sviluppare la malattia in forma grave, con interessamento oculare e del sistema nervoso centrale. L'evidenza di alterazioni del sistema immunitario (presenza di autoanticorpi, immunocomplessi circolanti, reazioni immunitarie anomale) permette di annoverarla tra le malattie autoimmuni.
I sintomi di questa malattia sono stati descritti per la prima volta dal medico greco Ippocrate nel 500 a.C., ma solo nel 1937 il medico turco Hulusi Behçet
considerò i tre sintomi principali, aftosi orale ricorrente, aftosi genitale ricorrenti e infiammazione dell'occhio (uveite)
UVEITI
come aspetti della stessa malattia. Multi fattoriali sono i sintomi della malattia, per cui il reumatologo (specialista preposto per questa patologia) deve spesso coordinarsi e confrontarsi con un'equipe di medici quali il dermatologo, l'oculista, il ginecologo, il gastroenterologo, il Neurologo e l'Odontoiatra.
MANIFESTAZIONI OCULARI
Si sono potute evidenziare grandi varietà di lesioni oculari , comprese iridocicliti anteriori, cataratta, glaucoma, coinvolgimento del segmento oculare posteriore con vasculite, vitreite, retinite, panuveite, edema retinico, degenerazione maculare citoide, occlusione venosa o arteriosa, edema discoide e/o distacco retinico.
L’uveite, la forma più frequentemente rilevata e complicata, viene classificata a seconda della localizzazione anatomica della flogosi:
UVEITI
ANTERIORE: irite o del corpo ciliare(ciclite);
POSTERIORE: della coroide e retina(uveite posteriore);
INTERMEDIA : della retina periferica e della parte piana del corpo ciliare(parsplanite);
PANUVEITE : flogosi generalizzata dell’intera uvea.
Il coinvolgimento oculare si verifica dal 43% al 72% dei pazienti e la riduzione significativa o perdita della vista è presente nel 25% di questi pazienti. Nel maschio l’alterazione visiva è più frequente e grave e nell’80% dei pazienti maschi si può evidenziare una malattia oculare bilaterale.
Il coinvolgimento oculare è raro come prima manifestazione di malattia (presente nel 10 – 13 % dei casi segnalati) ed è più frequente, come primo segno di MB, meno grave, nella femmina rispetto al maschio. Nel corso della malattia, però, il 75% dei pazienti presenta comunque coinvolgimento oculare, che, come già riferito, ha aspetti molto più gravi nell’uomo rispetto alla donna.
L’uveite anteriore, la più frequente, è tipicamente ricorrente e può risolversi spontaneamente. L’irite con ipoopion è stata la prima patologia oculare correlata alla MB descritta ed è anch’essa un segno clinico relativamente transitorio, poichè può risolversi spontaneamente in pochi giorni. Tra la comparsa di patologia oculare e cecità, nei soggetti non trattati, intercorrono solitamente circa 5 anni.. I vasi retinici si dilatano e diventano permeabili alla colorazione durante angiografia con fluoresceina e questo è un importante aspetto diagnostico negativo sulla evoluzione della malattia oculare in atto .
Nella fase infiammatoria acuta oculare della MB, si osserva una vasculite neutrofila, perivascolare e diffusa , dell’iride , del corpo ciliare e dei vasi coroidali.
Anche i vasi retinici sono infiltrati da polimorfonucleati.
Durante la remissione della MB e della malattia oculare ad essa correlata, si osserva una infiltrazione perivascolare di linfociti e plasmacellule nell’iride; nella retina si evidenzia infiltrazione sia di plasmacellule che di linfociti, mentre nella fase finale (cecità ) si evidenziano fibrosi ed occlusione vasale.
Le lesioni oculari sono una delle manifestazioni più gravi della Malattia di Behçet. Compaiono nelle fasi iniziali della malattia o comunque entro i primi 2 anni e ne rappresentano il sintomo di esordio in circa il 10-20% dei casi. La presentazione tipica è una panuveite recidivante con vasculite retinica occlusivo-necrotizzante, bilaterale nel 75% dei casi. L’ uveite anteriore si presenta come unica manifestazione solo in circa il 10% dei pazienti. Si tratta di un’ uveite non granulomatosa con o senza ipopion che può dare come complicanze: sinechie posteriori, cataratta, glaucoma secondario,edema maculare.
Più rara la presenza di sclerite o episclerite. La manifestazione oculare tipica della M. di Behçet è l’ uveite posteriore con infiammazione cellulare del corpo vitreo, presenza di essudati intraretinici con emorragie, ma soprattutto la vasculite occlusivo-necrotizzante del polo posteriore e della periferia retinica .
Un’altra manifestazione frequente è l’occlusione venosa di tipo trombotico con grave quadro emorragico retinico.
L’infiammazione della testa del nervo ottico è una caratteristica costante durante gli episodi infiammatori e può persistere nel tempo. La complicanza più comune negli occhi in cui persiste l’infiammazione è l’edema maculare cistoide, ma nei casi in cui è presente un edema retinico diffuso incontrollato si può manifestare anche un distacco di retina totale di tipo essudativo.
Fluorangiografia retinica che mostra edema del disco ottico, edema maculare e vasculite retinica.
L’occlusione vascolare determinando ipossia da luogo alla formazione di aree retiniche ischemiche con vasi collaterali di compenso, o a neovascolarizzazione del disco ottico con possibili emorragie vitreali. D’altro canto l’occlusione vascolare può determinare una completa avascolarità retinica che porta inevitabilmente ad un quadro terminale di atrofia del nervo ottico e degenerazione maculare.
TERAPIA
Secondo le raccomandazioni EULAR recentemente pubblicate, tutti i pazienti con malattia di Behçet ed interessamento del segmento posteriore dell’ occhio dovrebbero essere trattati con corticosteroidi (al dosaggio di 1 mg/kg/die) ed azatioprina (1-2 mg/kg/die); altre molecole di uso convenzionale sono la ciclosporina (3-5 mg/kg/die), clorambucil (0.1-0.2/kg/die), ciclofosfamide (1 gr/mese e.v.), Metotrexate (15-20 mg/sett. e.v.) e l’Interferon α (9 MU/sett.).
Altri farmaci sistemici
Corticosteroidi
Azatioprina
Clorambucil
Colchicina
Ciclofosfamide
Ciclosporina
Interferone-a
Talidomide
Methotrexate
Sulfasalazina
Micofenolato mofetil
Pentossifillina
Il TNFa rappresenta una citochina chiave nel processo patogenetico della malattia di Behçet, inoltre diversi elementi cellulari, tra cui le cellule T gd sono forti produttori di TNF-a. In tale logica appare razionale l’impiego terapeutico di farmaci anti-TNF-a (Infliximab, Adalimumab ed Etanercept).
L'Infliximab (INN; nome commerciale Remicade) è un anticorpo monoclonale chimerico usato come farmaco per trattare le malattie autoimmuni.Infliximab è stato approvato dalla Food and Drug Administration (FDA) per il trattamento della psoriasi, la malattia di Crohn, la spondilite anchilosante, artrite psoriasica, artrite reumatoide e la colite ulcerosa. Infliximab ha ottenuto la sua approvazione iniziale da parte della FDA per il trattamento della malattia di Crohn nell'agosto del 1998.
Infliximab è un anticorpo artificiale. È stato originariamente sviluppato nel topo con le normali tecniche per la produzione di anticorpi monoclonali. È stato poi modificato tramite tecniche di ingegneria genetica per renderlo simile ad un anticorpoumano in modo da ridurne l'immunogenicità. La modifica consiste nella sostituzione della porzione costante dell'anticorpo di topo con quella della IgG1 umana.
Infliximab è stato sviluppato da Junming Le e Jan Vilcek alla New York University School of Medicine ed è commercializzato negli Stati Uniti da Centocor Ortho Biotech (Centocor), in Giappone da Mitsubishi Tanabe Pharma, in Cina Janssen Xian, e Schering-Plough (ora parte di Merck & Co). Infliximab costa circa 16.000 euro all'anno per paziente
Un caro saluto
Prof.Duilio Siravo
siravo@supereva.it
http://drsiravoduilio.beepworld.it
Cell.:3385710585
PROF.DOTT. DUILIO SIRAVO
http://drsiravoduilio.beepworld.it